
What You Need to Know About Developing Software as a Medical Device
Software that meets the definition of a medical device, commonly referred to as Software as a Medical Device (SaMD), is subject to different requirements than software that does not meet this strict definition. In ICS’ new series What You Need to Know About Developing SaMD we explore the complexities of medical device software.
About Medical Device Software
There are certain things you should consider when developing software products. This includes user needs, development environment, infrastructure and security among others. However, if you are developing software that meets the definition of a medical device, the list of things to consider expands and the requirements you must address grows.
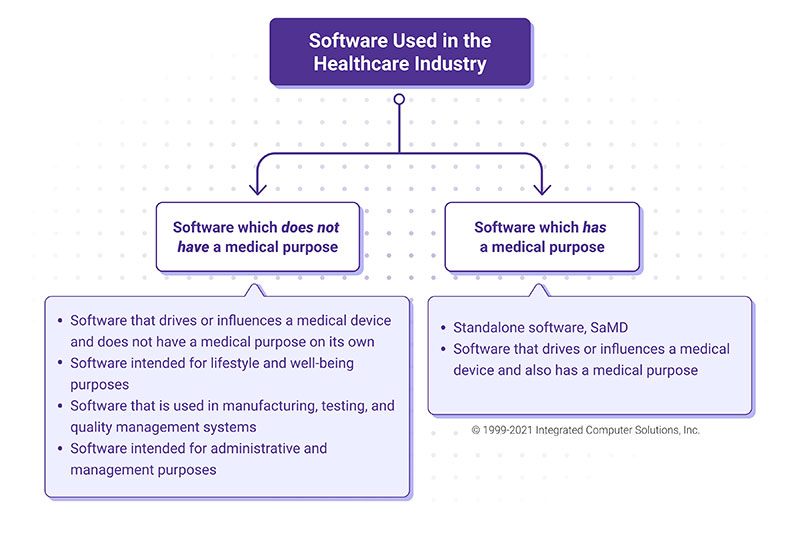
The software used in the healthcare industry can be divided into two groups: software that meets the definition of a medical device, and software that does not. Software that meets the definition of a medical device includes software intended for diagnosis, prevention, monitoring, prediction, prognosis, treatment or alleviation of disease or other conditions. Examples include apps that detect sleep apnea based on medical data; software that performs diagnostic image analysis for making treatment decisions in patients with acute stroke; and software that analyzes blood glucose levels and controls an insulin pump.
This type of software not only drives a medical device but also has a defined medical purpose. It can also encompass standalone software.
Software that does not meet the definition of a medical device includes lifestyle and wellbeing apps (e.g. apps aimed at helping users maintain a healthy lifestyle), apps that support administrative and management tasks (e.g. scheduling doctor’s visits, managing a quality system), software used in the manufacturing or testing of medical devices, and software that drives a medical device but does not have a medical purpose on its own. (This is covered by medical devices regulations either as a part/component of a device or as an accessory for a medical device.)
Regulatory Aspects of SaMDs
The International Medical Device Regulators (IMDRF) defines SaMD as Software intended to be used for one or more medical purposes that perform these purposes without being part of a hardware medical device.
At the outset of SaMD development, you need to address key regulatory aspects. The following are among the most fundamental — and most important — questions to answer:
- Does your software meet the definition of a medical device?
- What are the key regulations and standards that apply to your SaMD?
- What class is your SaMD?
- What regulatory pathway should you follow?
- What documentation should you develop?
In our series, we’ll address these questions, as well as explore mobile medical apps, AI-based SaMD, software design and development, and verification and validation
Regulations in the EU and U.S.
SaMD within the EU is regulated by the Regulation 2017/745 of the European Parliament and of the Council of 5 April 2017. Whereas medical device software in the U.S. is regulated by 21 CFR part 820. These regulations provide requirements concerning design, documentation, identification, production and surveillance of medical devices, among others.
However, because of the rapid pace of innovation and vast (and growing) number of software apps, the U.S. Food & Drug Administration (FDA) does not enforce requirements under the FD&C Act for all medical device software. The concept is consistent with the 21st Century Cures Act, which excluded certain software functions from the device definition.
The software functions outside of regulatory oversight include low-risk software that does not pose risk to the patient. This means that the manufacturer of such software is not required to submit premarket review applications or to list their software with the FDA. (Examples of software excluded from the FDA oversight can be found in the Policy for Device Software Functions and Mobile Medical Applications.)
Key Harmonized Standards
Apart from the regulations mentioned above, there are also standards and legal provisions that device makers should follow. For software, three standards are of particular importance: 62304, which describes software lifecycle processes; 62366, which specifies application of usability engineering to medical devices; and 14971, which is dedicated to risk management purposes.
Quality Management System
In addition to adhering to these standards and regulations, medical software manufacturers also must implement throughout their organizations a quality management system (QMS) or its specific parts. For instance, the ISO 13485 quality standard includes the requirements for a QMS and is recognized in the EU, Canada and Japan.
However, to enter the U.S. market you must meet the requirements of the quality system regulations (QSR) specified in the 21 CFR part 820. The requirements are similar to those outlined in ISO 13485. (To ease compliance, back in 2018 the FDA announced plans to harmonize its QSR with ISO 13485 but the effort has been delayed due to the pandemic.)
Guidance and Frameworks
To help stakeholders align with the regulations, the FDA in the U.S. and the MDCG in the EU provide guidance that includes further clarification of the regulatory requirements applicable to software. While not legally binding, this guidance will come in handy if you encounter a problem during software design and development.
Summary
While rewarding, developing SaMD is challenging and requires addressing myriad regulations. In the next installment in our series, we delve deeper into the regulatory aspects of SaMD, including software classification, pathways to market and key documents you should develop.
Learn more about ICS’ medical device and SaMD services.