Webinar Q&A
Q&A: Practical Advice for FDA’s 510(k) Requirements Webinar
Below are the questions asked by attendees during the live webinar on March 12 and the thought out answers from our experts. Please stay tuned for announcements on our future webinars that may address some of these topics.
Question: In case there are additional systems that are not under your control where your device interfaces with, what’s your risk management and design mitigation responsibility. Since this risk assumes bad actors, what level of systemic prevention and monitoring needs to be built in?
Answer: As we mentioned in the Q&A session of the webinar, the first approach, if possible, is to transfer risk. That is for your customer to create an environment that is secure or for them to manage the security of the other systems.
However transferring risk is not always possible. So another approach would be to use elements of Zero Trust. That would entail authenticating any device that communicates with your medical device and authenticating the data that is provided to make sure it had integrity (e.g., has not been tampered with.) You should also limit the access or prevent access entirely from the other devices to your device.
NIST has a document that provides information on Zero Trust. That document is NIST SP 800-207.
https://www.nist.gov/publications/zero-trust-architecture
A third approach would be: in your Threat Modeling and Risk Assessment, document all your assumptions and address the high risks that emerge from being in that environment with potentially bad actors. You could combine this approach with Zero Trust elements by considering Zero Trust mitigations to address risks identified.
Question: How much COTS (Commercial Off the Shelf) component information should be part of an SBOM, as we normally just use and not develop them ?
Apart from this, how much information should be shared in customer-facing SBoMs ?
Answer: Ideally, all the software that goes in your product will be reflected in the SBOM.
To get your vendors of COTS off-the-shelf software to provide SBOMs a collaboration with your purchasing team can be very helpful. They can require vendors to provide SBOMs as part of purchase agreements and help in general get SBOMs from vendors.
There can be an escalation , over a period of time, with vendors which include the following steps:
- Ask for the SBOM
- Prefer vendors that make SBOMs and make this known
- Only select vendors that provide SBOMs
The FDA requires a SBOM for a device that is looking for market access, but they do not require that it is provided to HDOs. (e.g., to your customers).
There are a couple of common concerns with providing SBOMs to customers but these could be addressed with an NDA:
- Competitive information could be disclosed
- Vulnerabilities can be identified by adversaries
Question: Is there a guideline on how many levels deep the SBOM should be?
Answer: Ideally, you should find all dependencies. But I would not recommend holding up a submission to the FDA because you are missing an "nth" level SBOM dependency.
If you don’t have SBOM information for one of your software components you should provide information as to why you don’t.
Question: Are there any recommended certifications that the FDA will accept as a valid certification that covers all their cybersecurity requirements?
Answer: No, I have not seen any certifications that cover all that the FDA is looking for. The FDA is taking the role of certifying. They want to see documentation for cybersecurity processes and artifacts (e.g. evidence such as a pentesting report) showing a full product lifecycle of security activities.
I do believe that the combination of IEC 81001-5-1 and AAMI SW96:2023 will give you the processes that you need that if followed (and fill in some gaps) will get you there.
Another path to consider is that there are many consulting companies that can help you prepare your cybersecurity submission and provice advice on closing any gaps.
Question: FDA guidance is also asking for 'Level of Support' and 'End-of-Support' data in SBOMs. Most tools don't support that. Do you have any suggestions for providing that in the machine readable SBOM?
Answer: SPDX is probably the most commonly used SBOM standard and 2.3 is the latest version.
“Level of Support” and “End of Support” are being considered for future revisions by the SPDX working group as you can see here: Add Software Level of Support property to Software Package · Issue #561 · spdx/spdx-3-model · GitHub
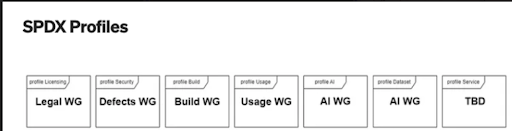
Being considered for the next version of SPDX (we don’t know i”Level of Support” and “End-of-Support” makes it in or not) is a number of profiles which include software license (Legal) and security (Defects) profiles (all machine readable). Here is the list of the profiles that are being considered.
Question: For Post market Vulnerability Evaluations, I consider system mitigations separately which will reduce my CVSS score from Critical to let's say medium. And then look for Remediation for whatever residual risk is left. But let's say in special use cases, for example, "Password Length to have complexity based on a particular length", how do we separate mitigation from remediation.
Answer: The wording can get confusing as mitigation, in particular, can be used to mean different things. In the context of a newly found post market vulnerability, time is a key differentiator between “mitigation” vs “remediation”. A “mitigation” (or the word I typically use is “containment”) should be implemented as quickly as possible and may not be a permanent solution. Maybe a “mitigation” could be applied in 24 hours. In my mind the “remediation” is the permanent solution that you can take time to develop because the “mitigation” is good enough until the “remediation” is ready to deploy.
To use a similar example, but slightly different, maybe it is Ok to allow single factor authentication, (with a strong password as you mention) for a period of months or even years until multi-factor authentication can be rolled out.
Question: How do you handle SBOM for embedded software?
Answer: As mentioned on the call if you are using Yocto, we have open-sourced a tool.
Zephyr (real time OS) has good SBOM support integrated.
You would have to check commercial SBOM tools to see how much support they have, but it is our experience that currently they don’t catch everything.
There can be a good amount of embedded software that is written for scratch, internally, so you can cover that by working with your internal developers.
For COTS embedded software, I’ll refer you to the answer above about getting your purchasing organization’s help to transition vendors to provide the SBOM information you need.
Question: Is there a significant difference between FDA and EU MDR/IVDR for software as a device?
Answer: This is an excellent question. The short answer is that cybersecurity requirements for SaMD are very similar between the FDA and EU MDR/IVDR. They are similar enough that one security process & effort can be executed that will mostly address requirements for both (but not all for both). Unfortunately there is not a single document or specification to reference. In fact there are four. The documents that you need to consider are:
- MDCG 2019-16 Guidance on Cybersecurity for Medical Devices
- FDA’s Cybersecurity In Medical Devices Quality Systems Considerations and Content of Premarket Submissions
- IMDRF Principles and Practices for Medical Device Cybersecurity
- IMDRF “Software as a Medical Device”: Possible Framework for Risk Categorization and Corresponding Considerations
Documents #1 and #2 are the guidance from medical device cybersecurity for the FDA and the EU.
The third document is a harmonization document for medical device cybersecurity across geographies. But don’t look at the document exclusively. It is helpful but it does not cover all requirements of #1 and #2.
Section 9.3, in particular of document #4 should be looked at for SaMD cybersecurity.
Question: Is it necessary to consider security associated with source code repository, malware on development and/or manufacturing computers, internal IT systems, etc.?
Answer: Your Secure Product Development Framework (SPDF) should say something about keeping your development environment safe. For example, section 5.1.2 in IEC 81001-5-1 has a paragraph about Development Environment security. It is only 2 sentences so it is very high level.
It can be helpful, but I would not say mandatory, if you can point to following ISO 27001 or SOC2 for your IT security.
Question: Penetration Testing, is it a hard requirement by FDA and/ or can it be done internally without certification of a third party?
Answer: Section V.C of the FDA’s Cybersecurity in Medical Device: Quality System Consideration and Content of Premarket Submissions is the section to review. It is the Cybersecurity Testing section. It says clearly that security testing reports should be submitted and as we showed on slide 12 there are 4 types of testing recommended by the FDA: Security Requirements (verification), Threat Mitigation, Vulnerability Testing, and Penetration testing.
As mentioned the pentesting can be done internally but it needs to be done by someone who has not worked on the design of the medical device.
A consideration of doing internal vs external pentesting is that it takes a special skill set. There are a number of external companies that offer competitive priced pentesting and even crowdsourced pentesting. We can recommend a few if needed.
No certification is required for pentesting. Just a test report. As another question mentions there are no standards for pentesting, for medical devices (JasPar for automotive in Japan is the only one that I know of) so no certification.
Question: After identifying a threat during the threat modeling, what's a good way to assign them a severity/priority? I know about CVSS but that is designed for scoring technical vulnerabilities, not threats. While a threat model may only output a threat, without a specific vulnerability.
Answer: There are two aspects to consider when quantifying threats and risks:
- Impact of the attack
- Feasibility of an attack (this is based on each of your threats)
You can put a number to those two aspects and come up with a risk score to rank risks .
Consider impact in terms of multiple levels of patient safety and assign a number to each. You may also want to consider operational availability of the device, financial, and data exfiltration impacts.
For feasibility you can use the categories of CVSS and assign a number from 1 to 5 for each. For example, the skill level required for the adversary to have to perform the attack can be rated from 1 to 5.
I’m going to point to an automotive cybersecurity standard because they have the best example of doing what I have described above. Take a look at appendix F, G, and H of ISO SAE 21434.
Appendix F shows an excellent approach to quantifying impact of an attack. It maps out four ratings levels for each category, and there are four categories: safety, operational availability, financial damage, and privacy (data).
Appendix G puts numbers to your threats to quantify feasibility. It even gives a second example based on qualifying CVSS.
Final appendix H pulls it all together with a simple example.
Question: Should these FDA regulations be applied on older devices (e.g. devices put on the market before these new cybersecurity regulations existed)?
Answer: “Bolting On” security is very difficult. If a medical device was not designed from the begining with security in mind, it is almost impossible to add the right level of security.
IMDRF has a document titled “Principles and Practices for the Cybersecurity of Legacy Medical Devices”. That is a good reference to securing legacy devices.
https://www.imdrf.org/working-groups/medical-device-cybersecurity-guide
Question: Are there additional Regulations/standards and/or "pathways" relevant for the UK mark
Answer: After Brexit; UK Conformity Assessed (UKCA) marking has replaced the CE mark in Great Britain (England, Wales, and Scotland). So the UK is in a process to transition to their own medical device legislation. It will be based on EU legislation but it is expected there will be differences.
There is a transition period for CE marked medical devices so that they may be placed on the market in Great Britain to the following timelines:
- General medical devices compliant with the EU MDD or EU AIMDD can be placed on the Great Britain market up until the sooner of expiry of certificate or 30 June 2028
- IVDs compliant with the EU IVDD can be placed on the Great Britain market up until the sooner of expiry of certificate or 30 June 2030
- General medical devices, including custom-made devices, compliant with the EU MDR and IVDs compliant with the EU IVDR can be placed on the Great Britain market up until 30 June 2030.
Devices in Northern Ireland continue to use the EU regulations and require CE marking (CE UKNI marking in case conformity assessment was done in UK) based on the Northern Ireland Protocol.
Question: What do you think is going to happen with the lack of standards for penetration testing for devices? Without a standard, there is a perverse incentive to do less testing in less depth so that fewer issues are found.
Answer: A very good question. Standards for pentesting would be very beneficial. The only one that I know of is JasPar which is a standard for automotive vulnerability testing in Japan.
https://www.jaspar.jp/standard_documents/detail_disclosure/495?select_tab=all
The FDA wants to see reports on the 4 types of security testing they recommend. So there will be some level of oversight on the quality of the testing.
For a pentest having a threat and risk analysis is very helpful. Provide it to the pentester so they know what you care most about and it will lead to a more efficient pentest. The threat and risk analysis can also be used as a scorecard as you should expect the pentest to have checked all attack surfaces and threats you listed.
Generally I believe white box pentesting (i.e., you provide information about the device under test to the pentester) is better because it is significantly more efficient and effective than black box pentesting (i.e., no information is given to the pentester)
Question: I know its one of the future options but can you clarify in a little more detail the security testing required against devices - what is the FDA wanting to see vs what they could decline? depth, approach? Its becoming critical to get this right for registration.
Answer: I look forward to presenting a webinar of FDA cyber-testing requirements. But an answer for now is:
- Test each individual security feature, (e.g. unit test) to make sure that is implemented correctly and meets the specification. I would call this verification.
- Test that the security controls meet your cybersecurity goals. I would call this validation.
- The FDA describes vulnerability testing as a number of approaches like fuzzing, scanning, testing attack surface, etc..
- Penetration testing is when there is someone independent from the design who thinks like an adversarial hacker and follow approaches they would.
The FDA wants to see reports on all four and they need to be tied back to your threat and risk analysis to show you have mitigated the highest risks.
Question: Are there clear definitions of what qualifies as a medical device, fitness device, healthcare app. There are many overlaps. Is there a category-wise listing?
Answer: A hardware device with software or a software app that a consumer could buy, needs to have an intended use for medical diagnosis or treatment for it to fall into the category of medical device.
If it is just being used for fitness, it would not be considered a medical device and would not need FDA approval.
A software app for a smartphone can fall into the category of “Software as a Medical Device” (SaMD) under the same definition: if it is being used for medical diagnosis or treatment.
Following are a couple of examples. You can see the intent is medical in nature (not just fitness).
https://news.samsung.com/us/fda-cleared-irregular-heart-rhythm-notification-for-galaxy-watch/
Question: In cases where a manufacturer submitted a 510(k) to address some high-risk vulnerabilities in a currently marketed device with an OS change but there are still high-risk vulnerabilities in the update, would the FDA reject the submission? This would of course delay getting the targeted mitigations out to the field.
Answer: FDA’s Postmarket Management of Cybersecurity in Medical Devices talks about controlled versus uncontrolled risks. I think the FDA’s acceptance of this change is going to depend if risks are controlled. I think if you can demonstrate the risks are controlled with this software update and a future update will address all remaining risks, and that plan is explained, I think that is a reasonable approach.
Question: Does vulnerability management apply to finding CVE's only, or should bugs also be considered? What would be an efficient way to get a list of bugs?
Answer: Some bugs can turn into vulnerabilities. The classic example is buffer overflows. So when bugs are being ranked to be fixed, the impact of the bug on cybersecurity should be considered. Classic software static and dynamic code analysis should be run along with good classic software testing approaches and this is expected by the FDA.
The four types of testing that we listed on slide 12 will also yield a list of bugs to fix.
Examples include:
- For verification an example would be a cryptographic algorithm is not implemented in a secure way. A fairly common mistake is not to use a true random number generator, that can't be predicted, when creating keys for symmetric encryption.
- For validation testing, an example would be the key length is not long enough to withstand brute force attacks of future compute platforms (e.g., quantum computers)
- For vulnerability testing, an example test is a TLS scan, and when run it may reveal self-signed certificates which ideally would be addressed.
- For pentesting, an example would be debug code is left in the production code and could give root privilege like control over the device.
Question: Can you please give me examples of FDA architecture views with sample diagrams and explanation of the ones below:
Global System View
Multi-Patient Harm View
Updatability/Patchability View
Security Use Case View(s)
Apart from this I need more information about Security Use Case View(s).
What exactly should these views talk about from operational states and how should they be shown Threat model or architecture ?
Answer: The approach that I like to take when threat modeling, which is a lead nice into risk analysis is to create Data Flow Diagrams (DFD). This entails creating a network diagram of the system that includes the network interfaces, and assets to be protected. This really helps to visualize the attack surface and to generate a list of threats. This approach also has the advantage that it gives you a view that is a really good starting point for the four views you list above (which the FDA is asking for in their pre-market submissions).
In our upcoming threat modeling and risk analysis webinar we’ll do a live exercise where we’ll create the DFD, identify threats, and rank risks. This will give an example that I think will be helpful.