
Algorithms, Data Quality Key to Safe AI-based Software as a Medical Device
Focusing on challenges related to developing AI-based Software as a Medical Device (SaMD), this blog explores current regulations and guidelines in the U.S and EU. For more on SaMD, join us at 1 pm EST on March 3 for a live webinar 5 Key Considerations at the Start of SaMD Development. We'll cover important aspects you should address at the outset of the SaMD development process to keep your project running smoothly, including regulatory concerns and technology considerations.
Artificial Intelligence (AI) is one of the greatest achievements of our times and is driving technological progress in many areas, including healthcare and medical devices. One of the reasons is the large number of potential applications and benefits of AI in medicine. For instance, these devices can detect rare health conditions, provide more accurate analysis of complex medical images and data, improve treatment outcomes for patients, and accelerate drug development.
In light of this potential, the number of approved AI-based medical devices has substantially increased over the last few years. As of January 2022, there are 343 FDA-approved AI-based medical devices in the U.S. [1] and 240 devices approved in the EU [2].
Sounds like a lot. But, the number is far below the potential for these devices. The reason: incorporating AI/machine learning (ML) within a medical device triggers an exceptionally thorough review process. Certainly, a high level of due diligence is warranted for devices that include AI and machine learning, which the FDA defines as “techniques used to design and train software algorithms to learn from and act on data.” However, there would exist many more approved devices – devices that can help improve patient outcomes – if the certification process was smoother and more clear cut.
Key AI-based SaMD Challenges
AI-based SaMD has the ability to learn from real-world data and change its performance. This capability makes them unique. In fact, it is considered one of the greatest benefits of AI. Still, it can pose significant risk to the patient.
Algorithm Type
Until now, FDA approved only algorithms that are “locked” prior to release to the field. The locked algorithms provide the same result each time the same input is provided and apply fixed functions. Changes to this kind of algorithm are made through a well-defined process, which is controlled by the manufacturer. Therefore the manufacturer assures that the system performs as intended and does not pose a risk to the patient.
On the other side, we’ve got adaptive algorithms, for instance continuously learning algorithms that change their behavior in the field using a defined learning process. This type of algorithm presents greater risk to the patients as the manufacturer is unable to fully control the algorithm modifications. As a result, they can present safety concerns once the device is released for use.
The challenge with both types of algorithms is that their modifications often involve algorithm architecture changes, re-training with new data sets, or clinical performance changes – all of which can be considered significant changes from a regulatory perspective. This means that the changes should be subjected to the premarket review process in the U.S under the software modifications guidance or to significant change notification and notified body assessment in the EU. Therefore, it’s crucial to reflect the dynamic nature of the AI SaMD in the current regulations and harmonized standards.
Data Quality
Another challenge that the AI SaMD manufacturers have to face is that the power of algorithms lies in the quality and quantity of data, and submissions may get rejected based on data quality.
To minimize this possibility device makers should considered several elements in this area, including:
- Amount of data points
- Representativeness of data points
- Whether data points match the SaMD’s intended use
- Source of data
- Errors in data labelling
- Overfitting or underfitting of data (data sets either align too closely or not closely enough with the requirements of the data models being used)
The other side of the coin is how to obtain the data. While we collect a vast amount of data, it is strictly protected and regulated by HIPAA in the U.S and GDPR in the EU. Thus it is difficult – sometimes impossible – to gain access to databases with relevant and diverse data.
Regulations
There are currently no harmonized standards specifically dedicated to AI-based medical devices in the U.S. and EU. Therefore, these products should meet the general regulatory requirements applicable to all medical device software, for instance EN 62304, MDR 2017/745, and 21 CFR part 820. (More information on the SaMD regulations can be found in the ICS’ series What You Need to Know About Developing SaMD.)
Despite the current lack of harmonized standards in this area, regulators are trying to keep pace with rapidly evolving technology and on occasion release frameworks and guidelines to follow in order to be compliant. In January 2021, the FDA issued an Artificial Intelligence/Machine Learning (AI/ML)-Based Software as a Medical Device (SaMD) Action Plan that outlines the agency's five-step action to approach the oversight of SaMD that incorporates AI.
Notified bodies in the EU have also made an attempt to address the AI-gap in the current regulations. TÜV SÜD released the white paper Artificial Intelligence in Medical Devices Verifying and Validating AI-based Medical Devices, which provides the criteria for AI-based device assessment and the compliance path. The white paper references the Requirements Checklist prepared by Germany’s Association of Notified Bodies for Medical Devices (IG-NB) for further details of the process-oriented approach.
There’s also the Proposal for a Regulation Lying Down Harmonised Rules on Artificial Intelligence (AI Act) prepared by the EU commission. It establishes rules for the development and placement on the market of AI systems (not only medical devices). It follows a proportionate risk-based approach and lays down a solid risk methodology to define high-risk AI systems.
In a nutshell, AI is considered to be a high-risk AI system when it is intended to be used as a safety component of a product or is itself a product covered by the EU harmonization legislation. Based on this classification, all software that qualifies as a medical device under the MDR or IVDR with an AI component will be classified as a high risk AI system and will have to meet special requirements outlined in the AI Act.
Process-oriented Approach
Guidelines and regulatory frameworks try to address the aforementioned challenges and propose a process-oriented approach to achieve safety and efficacy of AI-based SaMD. This encompasses all product development phases, such as research, design, development, data management and post-market surveillance. The concept, developed by the FDA, is called Total Product Life Cycle (TPLC). It aims to assure that ongoing improvements and changes are implemented in a controlled manner. The TPLC is based on four principles:
Principle one
Manufacturer establishes and maintains quality systems and good machine learning practice (GMPL). It encompasses:
- Relevance of available data to the clinical problem and current clinical practice
- Acquiring data in a consistent manner that aligns with the SaMD’s intended use and modification plan
- Appropriate separation of training, tuning and test datasets
- Appropriate level of transparency of the output and algorithms
Principle two
Manufacturer performs initial premarket assurance of safety and effectiveness (i.e. submits a predetermined change control plan during the pre-market review of an AI/ML-based SaMD). The plan includes:
- Software Pre-Specification (SPS), which describes “what” the manufacturer intends to change through learning
- Predetermined Algorithm Change Protocol (ACP), which states "how" the algorithm will learn. The components of the ACP are presented in the chart below.
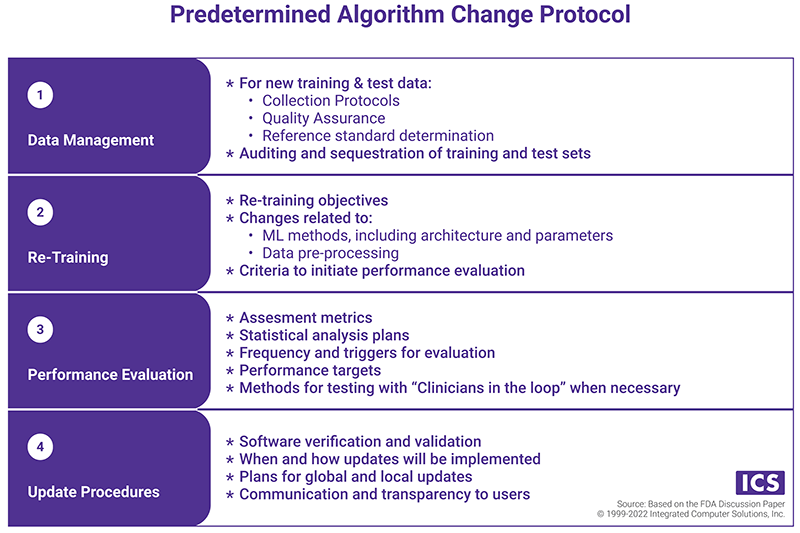
Principle three
Manufacturer introduces the modifications after the initial release based on the SPS and ACP.
- If the changes are within the scope of the SPS and the ACP, the manufacturer should document the change.
- If the modifications are outside of approved SPS and ACP, a new submission or focused FDA review may be required as outlined in the chart below.
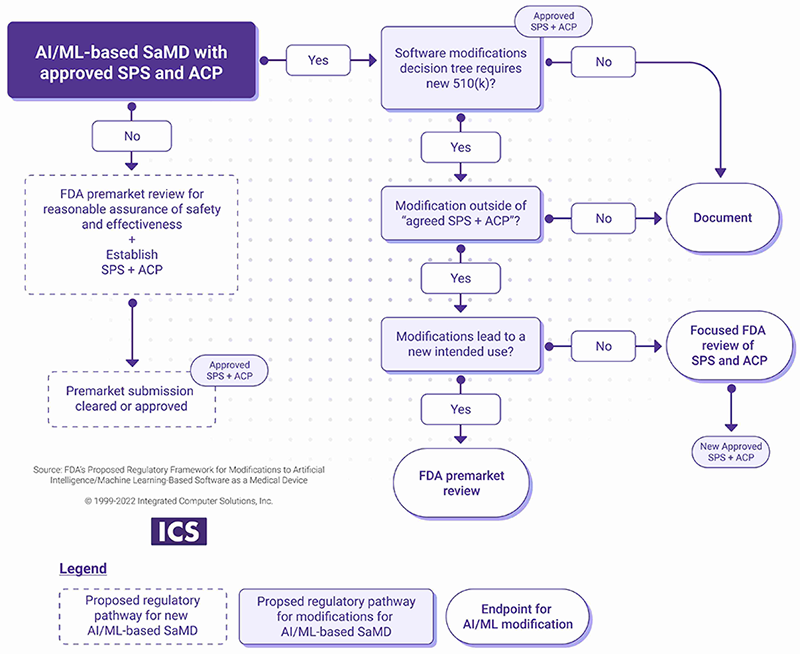
Principle four
Manufacturer monitors the real-world performance of AI-based SaMD and incorporates transparency with the regulators and users. This can be achieved through various mechanisms, for instance:
- Communication procedures that describe how users and FDA are notified of updates and their effects on performance
- Regular reporting on the performance of the SaMD on the market
EU regulators also emphasize the process-oriented approach, which extends beyond product planning and development and encompasses rigorous post-market surveillance (PMS) activities. The manufacturer should create a detailed PMS plan that describes how the manufacturer monitors changes in the performance and evaluates the impact on safety and efficacy of the AI-based SaMD. The Requirements Checklist, prepared by the IG-NB mentions Algorithm Change Protocol and SaMD Pre-Specifications as PMS plan elements.
The Takeaway
As regulators race to catch up with technological advancements, especially in the area of AI, they come up with new frameworks that SaMD manufacturers should follow. But technological progress routinely outpaces regulators’ ability to release new guidance, meaning regulations may fall behind the technology. Thus it is essential that medical device manufacturers exercise due diligence when developing and implementing changes to SaMD products.
References
[1] FDA, AI/ML-Enabled Medical Devices, https://www.fda.gov/medical-devices/software-medical-device-samd/artificial-intelligence-and-machine-learning-aiml-enabled-medical-devices
[2] Lancet Digital Health, 2021; 3: e195–203